Attivazione del legame carbonio-idrogeno
L'attivazione del legame carbonio-idrogeno o attivazione C-H è una reazione in cui viene rotto un legame carbonio-idrogeno.[1][2][3][4][5][6][7][8][9][10][11] Il termine è spesso limitato alle reazioni che coinvolgono complessi organometallici con trasformazioni che comprendono la coordinazione di un idrocarburo al metallo, o attraverso un "complesso alcano o arene" intermedio, o attraverso uno stato di transizione che porta ad un intermedio "M–C".[12][13][14] Ai fini della definizione è importante che il frammento idrocarburico rimanga associato nella sfera di coordinazione del complesso durante tutto l'evento che provoca l'attivazione C-H, in modo da essere influenzato dalla coordinazione al metallo.
Principi[modifica | modifica wikitesto]
I legami C-H sono tradizionalmente considerati non reattivi, ma sia studi teorici che ricerche sperimentali indicano che è possibile romperli sfruttando la coordinazione ad un metallo. Molta attività di ricerca si è occupata della progettazione e sintesi di nuovi reattivi e catalizzatori che possano influenzare l'attivazione C-H. Questi studi sono mirati al tentativo di sfruttare l'attivazione C-H sia per trasformare alcani abbondanti ed economici in pregiati composti organici funzionalizzati, e sia per modificare in modo efficiente la struttura di molecole complesse preesistenti.[15] In particolare, nel campo delle risorse energetiche sarebbe molto importante riuscire a trasformare in modo efficiente il metano (un gas abbondante ma scomodo da trasportare) in un combustibile liquido come il metanolo.[10][16]
Per l'attivazione selettiva di uno specifico legame C-H vengono utilizzati diversi concetti. Gruppi direzionanti sono usati per coordinare il metallo e quindi avvicinarvi il legame C-H desiderato. Gli eteroaromatici hanno una reattività intrinseca nei vari legami C-H (come ad esempio la posizione 2 del benzofurano). Inoltre, gli eteroatomi possono aumentare l'acidità di legami C–H adiacenti.
Nella funzionalizzazione si usa distinguere tra ossidazioni C–H, in cui un legame C-H viene trasformato in un legame C-eteroatomo, e reazioni di accoppiamento. In quest'ultimo caso si ottengono prodotti analoghi alle reazioni di accoppiamento incrociato. Tuttavia, qui il substrato organometallico prefunzionalizzato può sostituire il composto non funzionalizzato.
La maggior parte delle attivazioni C–H risultano al momento poco attraenti perché sono richieste condizioni piuttosto drastiche (alta temperatura, condizioni fortemente basiche o acide, ossidanti forti). Tuttavia la ricerca sta producendo condizioni di reazione sempre più blande, ampliando notevolmente l'ambito di applicabilità di questi processi.[17] Per effettuare reazioni di attivazione C–H si è iniziato a utilizzare anche l'organocatalisi; in determinate circostanze questo approccio può risultare più conveniente, dato che non vengono utilizzati metalli.[18]
Storia[modifica | modifica wikitesto]
La prima reazione di attivazione è a volte attribuita a Otto Dimroth, che nel 1902 descrisse la reazione tra benzene e l'acetato di mercurio(II), ma in genere questa non è considerata una vera attivazione C–H. Come osservato da Goldman e Goldberg,[13] l'attivazione C–H è per alcuni aspetti simile all'attivazione H–H, dato che entrambe si possono ottenere per addizione elettrofila o per addizione ossidativa.
Dal punto di vista della chimica organometallica moderna la prima vera attivazione C–H fu descritta nel 1965 da Joseph Chatt,[19] che ottenne l'inserzione di un atomo di rutenio coordinato a 1,2-bis(dimetilfosfino)etano in un legame C-H del naftalene. Nel 1969 Alexander E. Shilov riportò che il complesso K2PtCl4 induce scambio isotopico tra metano e acqua pesante, proponendo che il meccanismo prevedesse la coordinazione del metano al Pt(II). Nel 1972 il gruppo di Shilov riuscì a produrre metanolo e cloruro di metile da metano e acqua, usando quantità stechiometriche di K2PtCl4 o quantità catalitiche di K2PtCl6. Shilov lavorava e pubblicava i suoi risultati nell'Unione Sovietica ai tempi della guerra fredda, e per questo motivo il suo lavoro rimase sostanzialmente ignorato dagli scienziati occidentali. Il cosiddetto sistema di Shilov è ora riconosciuto come l'unico vero sistema catalitico per la funzionalizzazione di alcani.[13]
Dal punto di vista dell'addizione ossidativa, nel 1970 Malcolm Green descrisse l'inserzione fotochimica in un legame C–H del benzene da parte dell'atomo di tungsteno del complesso Cp2WH2,[20] e nel 1979 George M. Whitesides fu il primo ad ottenere una attivazione C–H alifatica intramolecolare.[21]
Il passo successivo fu descritto indipendentemente da due gruppi di ricerca nel 1982. R. G. Bergman descrisse la prima attivazione C–H di un idrocarburo totalmente saturo tramite addizione ossidativa con un metallo di transizione. La reazione fotochimica del complesso porta alla specie coordinativamente insatura Cp*Ir(PMe3), che dà quindi addizione ossidativa con cicloesano o neopentano formando i corrispondenti complessi alchilidruri Cp*Ir(PMe3)HR (R = cicloesile o neopentile).[22] W. A. G. Graham trovò che gli stessi idrocarburi reagiscono sotto irradiazione con Cp*Ir(CO)2 per dare i relativi alchilidruri Cp*Ir(CO)HR.[23] Nell'ultimo caso la reazione procede presumibilmente per addizione ossidativa all'intermedio a 16 elettroni di iridio(I), Cp*Ir(CO), formato per irradiazione di Cp*Ir(CO)2.

Attivazione C–H di Bergman et al. (asinistra) e Graham et al. (a destra)

Esempi[modifica | modifica wikitesto]
L'attivazione selettiva e la funzionalizzazione di legami C–H in alcani è stata descritta nel complesso di tungsteno Cp*W(NO)(η3-allyl)(CH2CMe3):[24]

Attivazione C–H del pentano come riportata da Baillie e Legzdins, 2014.

Nell'esempio illustrato l'alcano pentano è convertito selettivamente nell'idrocarburo alogenato 1-iodopentano. La trasformazione è effettuata per termolisi di Cp*W(NO)(η3-allyl)(CH2CMe3) a temperatura ambiente, che provoca l'eliminazione di neopentano in un processo di pseudo primo ordine. Si genera un intermedio non osservabile a 16 elettroni elettronicamente e coordinativamente insaturo contenente un legante η2-butadiene. In seguito, l'attivazione intermolecolare di una molecola di solvente pentano forma un complesso a 18 elettroni con un legante n-pentile, e in un processo successivo viene liberato 1-iodopentano per reazione con iodio a –60 °C.
Anche i legami C–H degli areni, benché piuttosto inerti, possono essere attivati con complessi metallici. Un esempio è l'accoppiamento olefinico di Murai.[25] Anche la reazione della figura successiva coinvolge una attivazione C–H; in questo caso un complesso di rutenio dà ciclometallazione con la N,N-dimetilbenzilammina:[26]
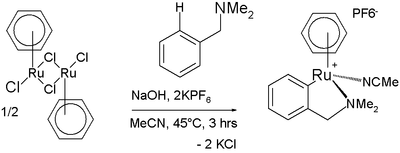
Ciclometallazione con una benzilammina sostituita

L'attivazione C–H di un alchene con un catalizzatore di rodio porta alla sintesi di una enamina biciclica tensionata:[27]

Attivazione C–H di Yotphan et al. 2008

Note[modifica | modifica wikitesto]
- ^ Crabtree 2001
- ^ Crabtree 2004
- ^ Lersch e Tilset 2005
- ^ Vedernikov 2007
- ^ Davies e Manning 2008
- ^ Boutadla 2009
- ^ Balcells et al. 2010
- ^ Lyons e Sanford 2010
- ^ Shul’pin 2010
- ^ a b Hashiguchi et al. 2012
- ^ Kuhl et al. 2012
- ^ Arndtsen et al. 1995
- ^ a b c Goldman e Goldberg 2004
- ^ Periana et al. 2004
- ^ Wencel-Delord e Glorius 2013
- ^ Crabtree 2010
- ^ Wencel-Delord et al. 2011
- ^ Pan 2012
- ^ Chatt e Davidson 1965
- ^ Green e Knowles 1970
- ^ Foley e Whitesides 1979
- ^ Janowicz e Bergman 1982
- ^ Hoyano e Graham 1982
- ^ Baillie e Legzdins 2014
- ^ Murai et al. 1993
- ^ Chetcuti e Ritleng 2007
- ^ Yotphan et al. 2008
Bibliografia[modifica | modifica wikitesto]
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley e T. H. Peterson, Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution, in Acc. Chem. Res., vol. 28, n. 3, 1995, pp. 154–162, DOI:10.1021/ar00051a009. URL consultato il 26 marzo 2014.
- R. A. Baillie e P. Legzdins, Distinctive Activation and Functionalization of Hydrocarbon C–H Bonds Initiated by Cp*W(NO)(η3-allyl)(CH2CMe3) Complexes, in Acc. Chem. Res., vol. 47, n. 2, 2014, pp. 330–340, DOI:10.1021/ar400108p. URL consultato il 31 marzo 2014.
- D. Balcells, E. Clot, O. Eisenstein, C–H Bond Activation in Transition Metal Species from a Computational Perspective, in Chem. Rev., vol. 110, n. 2, 2010, pp. 749–823, DOI:10.1021/cr900315k. URL consultato il 26 marzo 2014.
- Y. Boutadla, D. L. Davies, S. A. Macgregor, A. I. Poblador-Bahamonde, Mechanisms of C–H bond activation: rich synergy between computation and experiment, in Dalton Trans., 2009, pp. 5820–5831, DOI:10.1039/B904967C. URL consultato il 26 marzo 2014.
- J. Chatt e J. M. Davidson, 154. The tautomerism of arene and ditertiary phosphine complexes of ruthenium(0), and the preparation of new types of hydrido-complexes of ruthenium(II), in J. Chem. Soc., 1965, pp. 843-855, DOI:10.1039/JR9650000843. URL consultato il 24 marzo 2014.
- M. J. Chetcuti e V. Ritleng, Formation of a Ruthenium–Arene Complex, Cyclometallation with a Substituted Benzylamine, and Insertion of an Alkyne, in J. Chem. Educ., vol. 84, n. 6, 2007, p. 1014, DOI:10.1021/ed084p1014. URL consultato il 31 marzo 2014.
- R. H. Crabtree, Alkane C–H activation and functionalization with homogeneous transition metal catalysts: a century of progress – a new millennium in prospect, in J. Chem. Soc., Dalton Trans., 2001, pp. 2437–2450, DOI:10.1039/B103147N. URL consultato il 24 marzo 2014.
- R. H. Crabtree, Organometallic alkane CH activation, in J. Organomet. Chem., vol. 689, n. 24, 2004, pp. 4083–4091, DOI:10.1016/j.jorganchem.2004.07.034. URL consultato il 24 marzo 2014.
- R. H. Crabtree, Introduction to Selective Functionalization of C−H Bonds, in Chem. Rev., vol. 110, n. 2, 2010, pp. 575–575, DOI:10.1021/cr900388d. URL consultato il 31 marzo 2014.
- H. M. L. Davies, J. R. Manning, Catalytic C–H functionalization by metalcarbenoid and nitrenoid insertion, in Nature, vol. 451, 2008, pp. 417–424, DOI:10.1038/nature06485. URL consultato il 26 marzo 2014.
- P. Foley e G. M. Whitesides, Thermal generation of bis(triethylphosphine)-3,3-dimethylplatinacyclobutane from dineopentylbis(triethylphosphine)platinum(II), in J. Am. Chem. Soc., vol. 101, n. 10, 1979, pp. 2732–2733, DOI:10.1021/ja00504a041. URL consultato il 26 marzo 2014.
- A. S. Goldman e K. I. Goldberg, Organometallic C–H Bond Activation: An Introduction, in ACS Symposium Series, 885, Activation and Functionalization of C–H Bonds, 2004, pp. 1-43.
- M. L. Green e P. J. Knowles, Formation of a tangsten phenyl hydride derivatives from benzene, in J. Chem. Soc. D, n. 24, 1970, pp. 1677–1677, DOI:10.1039/C29700001677. URL consultato il 26 marzo 2014.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick, R. A. Periana, Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction, in Acc. Chem. Res., vol. 45, n. 6, 2012, pp. 885–898, DOI:10.1021/ar200250r. URL consultato il 24 marzo 2014.
- James K. Hoyano e W. A. G. Graham, Oxidative addition of the carbon–hydrogen bonds of neopentane and cyclohexane to a photochemically generated iridium(I) complex, in J. Am. Chem. Soc., vol. 104, n. 13, 1982, pp. 3723–3725, DOI:10.1021/ja00377a032. URL consultato il 26 marzo 2014.
- A. H. Janowicz e R. G. Bergman, Carbon–hydrogen activation in saturated hydrocarbons: direct observation of M + R−H → M(R)(H), in J. Am. Chem. Soc., vol. 104, n. 1, 1982, pp. 352–354, DOI:10.1021/ja00365a091. URL consultato il 26 marzo 2014.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord, F. Glorius, Beyond Directing Groups: Transition Metal-Catalyzed C–H Activation of Simple Arenes, in Angew. Chem. Int. Ed., vol. 51, n. 41, 2012, pp. 10236–10254, DOI:10.1002/anie.201203269. URL consultato il 26 marzo 2014.
- M. Lersch, M.Tilset, Mechanistic Aspects of C−H Activation by Pt Complexes, in Chem. Rev., vol. 105, n. 6, 2005, pp. 2471–2526, DOI:10.1021/cr030710y. URL consultato il 24 marzo 2014.
- T. W. Lyons, M. S. Sanford, Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions, in Chem. Rev., vol. 110, n. 2, 2010, pp. 1147–1169, DOI:10.1021/cr900184e. URL consultato il 26 marzo 2014.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda, N. Chatani, Efficient catalytic addition of aromatic carbon–hydrogen bonds to olefins, in Nature, vol. 366, 1993, pp. 529–531, DOI:10.1038/366529a0. URL consultato il 31 marzo 2014.
- S. C. Pan, Organocatalytic C–H activation reactions, in Beilstein J. Org. Chem., vol. 8, 2012, pp. 1374–1384, DOI:10.3762/bjoc.8.159. URL consultato il 26 marzo 2014.
- R. A. Periana, G. Bhalla, W. J. Tenn III, K. J. H. Young, X. Y. Liu, O. Mironov, C. Jones e V. R. Ziatdinov, Perspectives on some challenges and approaches for developing the next generation of selective, low temperature, oxidation catalysts for alkane hydroxylation based on the C–H activation reaction, in J. Mol. Catal. A: Chemical, vol. 220, n. 1, 2004, pp. 7-25, DOI:10.1016/j.molcata.2004.05.036. URL consultato il 26 marzo 2014.
- G. B. Shul’pin, Selectivity enhancement in functionalization of C–H bonds: A review, in Org. Biomol. Chem., vol. 8, n. 19, 2010, pp. 4217–4228, DOI:10.1039/c004223d. URL consultato il 26 marzo 2014.
- A. N. Vedernikov, Recent Advances in the Platinum-mediated CH Bond Functionalization, in Curr. Org. Chem., vol. 11, n. 16, 2007, pp. 1401–1416, DOI:10.2174/138527207782418708. URL consultato il 24 marzo 2014.
- J. Wencel-Delord, T. Dröge, F. Liu e F. Glorius, Towards Mild Metal-Catalyzed C–H Bond Activation, in Chem. Soc. Rev., vol. 40, n. 9, 2011, pp. 4740–4761, DOI:10.1039/C1CS15083A. URL consultato il 26 marzo 2014.
- J. Wencel-Delord e F. Glorius, C–H bond activation enables the rapid construction and late-stage diversification of functional molecules, in Nature Chem., vol. 5, 2013, pp. 369–375, DOI:10.1038/nchem.1607. URL consultato il 26 marzo 2014.
- S. Yotphan, R. G. Bergman e J. A. Ellman, The Stereoselective Formation of Bicyclic Enamines with Bridgehead Unsaturation via Tandem C–H Bond Activation/Alkenylation/ Electrocyclization, in J. Am. Chem. Soc., vol. 130, n. 8, 2008, pp. 2452–2453, DOI:10.1021/ja710981b. URL consultato il 31 marzo 2014.
Altri progetti[modifica | modifica wikitesto]
 Wikimedia Commons contiene immagini o altri file su Attivazione del legame carbonio-idrogeno
Wikimedia Commons contiene immagini o altri file su Attivazione del legame carbonio-idrogeno